Агенезия мозолистого тела головного мозга на узи у новорожденного
1. Сокращения:
• Агенезия мозолистого тела (АМТ)
2. Определения:
• Аксоны не пересекают среднюю линию и не образуют мозолистое тело:
о Агенезия: полное отсутствие мозолистого тела
о Гипогенезия: частичное или незавершенное формирование мозолистого тела:
— Встречается чаще, чем агенезия
о Дисгенезия: дефект развития мозолистого тела (например, пороки развития мозолистого тела при ГПЭ)
б) Лучевая диагностика:
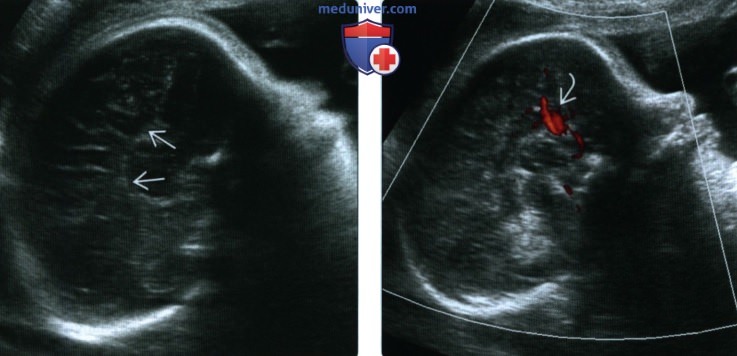 (Слева) При УЗИ в сагиттальной плоскости выявляется АМТ, отсутствие поясной извилины, видны радиально расположенные извилины, сходящиеся к III желудочку
(Слева) При УЗИ в сагиттальной плоскости выявляется АМТ, отсутствие поясной извилины, видны радиально расположенные извилины, сходящиеся к III желудочку
(Справа) При допплеровском УЗИ обнаруживается патологическое направление околомозолистой артерии, в норме идущей в переднезаднем направлении вдоль поясной извилины. Этот признак может помочь подтвердить диагноз АМТ.
2. УЗИ при агенезии, дисгенезии мозолистого тела головного мозга плода:
• УЗИ в режиме серой шкалы:
о Часто первым признаком является незначительная вентрикуломегалия
о Кольпоцефалия: расширение треугольников и затылочных рогов
• Желудочки в форме слезы
• ЦДК:
о Извилистость передних мозговых артерий
о Иногда выявляется непарная передняя мозговая артерия
о Околомозолистая артерия имеет патологическое направление
• В 50% случаев обнаруживаются другие пороки развития ЦНС:
о Чаще всего выявляются следующие пороки: СДУ, мальформация Киари 2-го типа (мальформация Арнольда-Киари), пороки, связанные с нарушением миграции/организации нейронов, энцефалоцеле, аномалии развития лицевого черепа по средней линии
о Нарушения строения других спаек конечного мозга
о МПК/комплекс AVID (англ. Asymmetric ventriculomegaly, interhemispheric cyst, dysgenesis of corpus calosum):
— Асимметричная вентрикуломегалия с МПК и дисгенезией мозолистого тела
— Киста может сообщаться с полостью желудочка или нет
о Межполушарные липомы:
— Гиперэхогенные новообразования, располагающиеся по средней линии, часто с признаками кальцификации
— В 50% случаев липомы сопровождаются АМТ
о Гетеротопии и пороки развития извилин
о Микроцефалия
• Пороки развития тела (АМТ встречается не только в сочетании с пороками развития ЦНС):
о Пороки сердца
о Врожденные диафрагмальные грыжи (ВДГ)
о Пороки развития ЖКТ и мочеполовой системы
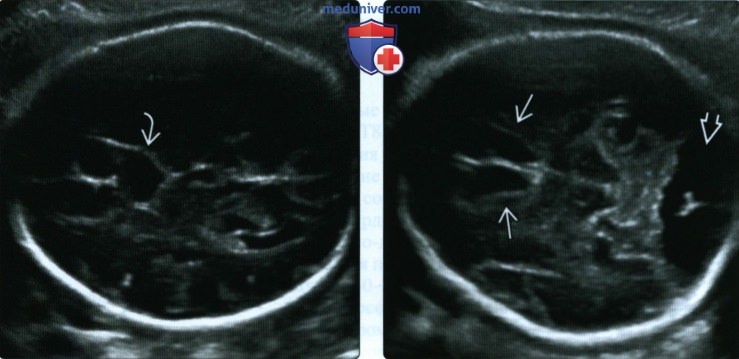 (Слева) При УЗИ у плода на 35-й неделе гестации сзади от предполагаемого места расположения ППП обнаруживается анэхогенная киста. При аномалиях развития мозолистого тела часто имеются МПК. Последние не следует путать с ППП, в норме располагающейся между передними рогами боковых желудочков.
(Слева) При УЗИ у плода на 35-й неделе гестации сзади от предполагаемого места расположения ППП обнаруживается анэхогенная киста. При аномалиях развития мозолистого тела часто имеются МПК. Последние не следует путать с ППП, в норме располагающейся между передними рогами боковых желудочков.
(Справа) Спереди от кисты передние рога имеют форму щели, ППП отсутствует. Также видно РБЦ. Амниоцентез подтвердил диагноз Т18.
3. МРТ при агенезии, дисгенезии мозолистого тела головного мозга плода:
• Т2-ВИ: картина, аналогичная выявляемой при УЗИ, но признаки более очевидны:
о Сагиттальная плоскость:
— Отсутствие мозолистого тела и поясной извилины
— Патологическое строение радиально расположенных извилин, сходящихся к III желудочку («солнечные лучи», или «спицы колеса»)
о Могут быть видны продольные пучки волокон мозолистого тела:
— Непересекающиеся волокна спайки, в норме образующие мозолистое тело, идут в переднезаднем направлении и не пересекают среднюю линию
— Зазубренная медиальная стенка боковых желудочков придает им форму полумесяца. Во фронтальной плоскости передние рога боковых желудочков имеют форму рогов быка
4. Рекомендации по лучевой диагностике:
• Основной метод исследования:
о Обычно диагноз ставят на основании данных УЗИ, однако в план стандартного обследования также рекомендуется включать МРТ плода:
— В половине случаев в пренатальном периоде МРТ позволяет обнаружить пороки развития, пропущенные при УЗИ:
Многие из них, например гетеротопия серого вещества, слишком малы и не могут быть выявлены при УЗИ
— Часто МРТ применяют в случаях, когда проведение УЗИ представляет трудности (ожирение у матери и олиго-гидрамнион)
• Советы по проведению исследования:
о Тщательно выполненное УЗИ позволяет точно поставить правильный диагноз:
— АМТ часто не замечают или путают с гидроцефалией, в особенности во II триместре
— При головном предлежании плода оптимально проводить ТВУЗИ
о Следует провести диагностику сопутствующих пороков развития головного мозга и тела плода
о Проводят МРТ в трех ортогональных плоскостях, в режиме тонких срезов с использованием импульсной последовательности HASTE (SSFSE):
— Во многих случаях МРТ в сагиттальной и фронтальной плоскостях позволяет получить больше данных, чем исследование в стандартной аксиальной плоскости
в) Дифференциальная диагностика агенезии, дисгенезии мозолистого тела головного мозга плода:
1. Незначительная вентрикуломегалия:
• Нормальное строение желудочков, наличие ПИП, нормальное строение извилин
2. Лобарная ГПЭ:
• Серп отсутствует, или его строение не соответствует норме
• Отсутствие ППП, слияние передних рогов, слияние столбов свода мозга, слияние долей таламуса
3. Септооптическая дисплазия:
• Отсутствие ППП, слияние передних рогов:
о Передние рога уплощенной или квадратной формы
• Мозолистое тело сформировано, но истончено
4. Очаговое поражение мозолистого тела:
• Волокна мозолистого тела разрушены в необычных местах
г) Патологоанатомические особенности. Общие сведения:
• Генетика:
о Чаще всего имеет спорадический характер
о За формирование мозолистого тела отвечают многие гены. Таким образом, его нарушение может происходить на разных этапах
о Хромосомные аномалии составляют 10-20%:
— Т18, Т13, Т8
— Триплоидия
• Эмбриогенез:
о Мозолистое тело формируется между 8-й и 20-й неделями гестации по средней линии из зародышевого листка
— Оно продолжает утолщаться и после рождения
о Мозолистое тело в основном формируется в переднезаднем направлении:
— В таком порядке: задняя часть колена/передняя часть ствола → передняя часть колена/задняя часть ствола → валик → клюв
д) Клинические особенности:
1. Клиническая картина:
• Самые частые субъективные и объективные симптомы:
о Незначительная вентрикуломегалия:
— В 3% случаев при незначительной вентрикуломегалии имеется АМТ
2. Демографические особенности:
• Эпидемиология:
о Частота возникновения в общей популяции 0,3-0,7%
3. Естественное течение и прогноз:
• Изолированная АМТ:
о В 75% случаев к 3-му году жизни развитие нормальное или близкое к нормальному
• При сочетании с другими пороками развития, при наличии синдромов или хромосомных нарушений прогноз неблагоприятный
• Приблизительно в 15% случаев, которые расценивают как изолированные, сопутствующие пороки обнаруживаются уже после рождения
4. Лечение:
• Если находка признана изолированной, рекомендуется исследование кариотипа
е) Особенности диагностики. Признаки, учитываемые при интерпретации результатов:
• Диагноз изолированной АМТ трудно подтвердить до 20-22-й недели гестации:
о Патологию можно не заметить или спутать с гидроцефалией даже на поздних сроках беременности
• МРТ позволяет поставить правильный диагноз и выявить сопутствующие пороки развития
ж) Список использованной литературы:
1. Craven I et al: Antenatal diagnosis of agenesis of the corpus callosum. Clin Radiol. 70(3):248-53, 2015
2. Riiland AM et al: Prenatal diagnosis of anomalies of the corpus callosum over a 13-year period. Ultraschall Med. ePub, 2015
3. Noguchi R et al: Outcomes of patients with prenatally diagnosed agenesis of the corpus callosum in conjunction with ventriculomegaly. Arch Gynecol Obstet. 290(2):237-42, 2014
4. Paladini D et al: Agenesis of the fetal corpus callosum: sonographic signs change with advancing gestational age. Ultrasound Obstet Gynecol. 42(6):687-90, 2013
Видео УЗИ головного мозга плода в норме
Редактор: Искандер Милевски. Дата обновления публикации: 17.9.2021
Агенезия мозолистого тела
Агенезия мозолистого тела — это врожденное отсутствие мозолистого тела либо его части. Аномалия обусловлена генетическими нарушениями, сосудистыми мальформациями, тератогенными факторами. Основные признаки заболевания: двигательные расстройства, задержка психоречевого развития, судорожные приступы. При негрубом (частичном) варианте патологии возможно малосимптомное течение. Для диагностики состояния назначается церебральные КТ или МРТ, нейросонография у новорожденных, генетические исследования. Лечение симптоматическое: медикаментозная коррекция осложнений, реабилитационные программы.
МКБ-10
Общие сведения
Агенезия мозолистого тела (АМТ) — один из наиболее частых пороков нервной системы. Распространенность болезни в популяции составляет от 0,05% до 7% среди новорожденных, причем в группе детей с замедленным становлением психики агенезия встречается у 2,3%. Калифорнийская программа по изучению врожденных пороков предоставляет другие данные по частоте агенезии — 1,4 на 10000 живых новорожденных. Впервые состояние было описано в 1812 году в ходе аутопсии, проведенной немецким анатомом И. Рэйлем, и названо «природной моделью рассеченного мозга».
Причины
Точные этиологические факторы заболевания не установлены. В современной неврологии преобладает мультифакториальная теория, согласно которой для формирования врожденного порока ЦНС требуется комбинация неблагоприятных экзогенных и эндогенных причин. Ученые выделяют несколько наиболее вероятных предпосылок развития агенезии:
Основным фактором риска выступает недоношенность. У новорожденных, родившихся до 27-недельного срока гестации МТ истончено в задних отделах, между 28 и 30 неделями — только в области валика. У рожденных после 30 недели в неонатальном периоде изменения не обнаруживаются, хотя при нейропсихологическом исследовании у школьников зачастую выявляется дефицит межполушарной передачи познавательной информации.
Патогенез
Мозолистое тело (МТ) представляет собой крупный пучок комиссуральных нервных волокон. Это важный аксональный путь, который соединяет соответствующие зоны коры правого и левого полушарий. Анатомическая структура имеет длину 7-9 см, состоит из более 300 млн. аксонов. Формирование МТ начинается на этапе позднего нейроонтогенеза (8-9 недели эмбриогенеза). Его созревание в норме продолжается до 20-25 лет.
Агенезия возникает при нарушении дифференциации нервной трубки в период со 2 до 5 месяца внутриутробного развития. При полном отсутствии МТ третий мозговой желудочек остается открытым, не формируются столбы свода мозга, отсутствуют прозрачные перегородки. В 60% случаев при АМТ передней комиссуры нет вообще. В 10% она увеличена и берет на себя часть функций мозолистого тела у новорожденных, а также на следующих этапах постнатального периода.
Характерным анатомическим изменением является колпоцефалия, при которой расширены задние отделы боковых церебральных желудочков. Состояние не относится к истинной гидроцефалии новорожденных, а обусловлено уменьшением кортикальных ассоциативных путей. Еще один типичный признак порока — пучки Пробста, представляющие собой неправильно ориентированные аксоны, расположенные параллельно межполушарной щели.
Классификация
В практической неврологии состояние подразделяют на тотальное, когда орган полностью отсутствует, и частичное (парциальное), при котором визуализационные методы не обнаруживают отдельные участки МТ. Это имеет решающее значение для тяжести клинической картины, возможных осложнений. В соответствии с патогенетическими особенностями формирования врожденных пороков, выделяют следующие 3 формы болезни:
Симптомы
Клиническая картина агенезии мозолистого тела широко варьирует от практически бессимптомных форм (при гипоплазии) до критических нервно-психических расстройств при его грубом недоразвитии, сопровождающемся другими врожденными пороками ЦНС. У новорожденных признаки патологии могут вовсе отсутствовать и проявляться по мере взросления младенца задержкой психомоторного развития.
Двигательные нарушения определяются у 35-40% пациентов. Они проявляются мышечной гипотонией или дистонией, гипер- или гипорефлексией, нарушением глотательного и сосательного рефлексов. Дети позже начинают держать голову, испытывают затруднения при обучении сидению, ползанию, ходьбе. Могут отмечаться координационные нарушения, неуклюжая походка. Из пароксизмальных расстройств у новорожденных и детей первого года жизни преобладают судороги.
Мозолистое тело поддерживает связь между церебральными зонами, формирует межполушарную организацию высших психических процессов. При его агенезии либо гипоплазии у детей выявляются когнитивные расстройства. У новорожденных пациентов и в раннем детстве наблюдается задержка речи, снижение динамического компонента игровой деятельности. В дошкольном и школьном возрасте возникают проблемы с концентрацией внимания, расстройства памяти, при тотальной АМТ снижен коэффициент интеллекта.
Осложнения
Около 65% случаев заболевания сопровождаются сопутствующими врожденными патологиями, среди которых преобладают мальформации кортикального развития (22,8%), межполушарные кисты (14,3%), голопрозэнцефалия (14,3%). К более редким сопутствующим аномалиям относят кисты и гипоплазию мозжечка, синдром Арнольда-Киари. До 20% новорожденных, кроме структур ЦНС, имеют пороки нескольких внутренних органов.
У 75% больных с тотальным поражением наблюдается симптоматическая эпилепсия височно-лобной локализации, в 66% случаев выражены когнитивные нарушения. У 16% пациентов формируются расстройства аутистического спектра. Изредка встречаются патологии органа зрения в виде хориоретинальных лакунарных очагов, сочетанной аномалии зрительных нервов.
Диагностика
В качестве первичного метода обследования в пренатальном периоде проводится акушерское УЗИ. У новорожденных для скрининговой диагностики используется нейросонография, однако этот метод не всегда показывает хорошую информативность, особенно при парциальной агенезии. Для верификации диагноза назначаются следующие методы исследования:
Лечение агенезии мозолистого тела
Специфическая терапия отсутствует. Медикаментозное лечение назначается неонатологом или педиатром индивидуально с учетом ведущих патологических синдромов: у новорожденных, детей раннего возраста используются антиконвульсанты, нейрометаболические препараты, дегидратационная терапия. Основу медицинской помощи составляет комплексная реабилитация, которая включает следующие составляющие:
Прогноз и профилактика
Прогноз определяется видом врожденной аномалии мозолистого тела, наличием сопутствующих пороков развития ЦНС. Благоприятный исход наблюдается при частичной гипоплазии МТ, а в случае комбинированных церебральных пороков у новорожденных могут быть жизнеугрожающие осложнения. Профилактические меры включают медико-генетическое консультирование, исключение тератогенных влияний в гестационном периоде.
Недоразвитие мозолистого тела (агенезия)
Развитие головного мозга ребенка начинается внутриутробно и активно продолжается после рождения.
По исследованиям физиологов правое полушарие головного мозга – гуманитарное, образное, творческое – отвечает за тело, координацию движений, баланс, пространственное зрительное и кинестетическое восприятие.
Левое полушарие головного мозга – математическое, знаковое, речевое, логическое, аналитическое – отвечает за восприятие – слуховой информации, постановку целей и построений программ.
Единство мозга складывается из деятельности двух полушарий, тесно связанных между собой системой нервных волокон (мозолистое тело).
Мозолистое тело (межполушарные связи) находится между полушариями головного мозга в теменно-затылочной части и состоит из двухсот миллионов нервных волокон. Оно необходимо для координации работы мозга и передачи информации из одного полушария в другое.
Агенезия (нарушение, недоразвитие) мозолистого тела искажает познавательную деятельность детей. Если нарушается проводимость через мозолистое тело, то ведущее полушарие берет на себя большую нагрузку, а другое блокируется. Оба полушарие начинают работать без связи.
Нарушаются пространственная ориентация, баланс, осознание собственного тела, адекватное эмоциональное реагирование, координация работы зрительного и аудиального восприятия с работой пишущей руки.
Ребенок с такими проблемами не ползает, тяжело начинает ходить, с большим трудом начинает читать и писать, воспринимая информацию на слух или зрительно. У детей с данной патологией, если вовремя не начать коррекцию и последующую реабилитацию, возникает целый ряд серьезных проблем, которые являются серьезным препятствием в развитии и обучении, в том числе и школьном.
В том случае, если агенезия мозолистого тела не сопровождается никакими другими патологиями развития, прогноз для больного достаточно благоприятный. Примерно восемьдесят с лишним процентов таких детей развиваются практически без нарушений либо с пограничными проблемами в неврологическом развитии. Нужно признать, что главная «опасность» этого нарушения таится в том, что у ребенка не происходит закрепления полученных умений и навыков навсегда, часто случаются «откаты», ребенок требует всё время поддерживающей терапии с нарастающей нагрузкой для мозга. Такой подход должен сохраняться до 12-14 лет ребенка, пока межполушарные связи окончательно не сформируются. Форсировать события здесь, к сожалению, нельзя. Иначе не избежать сочетанных проблем и других патологических состояний, которые усиливают симптоматику и ухудшают клиническую картину.
Агенезия мозолистого тела, хоть и встречается сравнительно часто, тем не менее, является малоизученным состоянием, особенно на просторах нашей страны.
Гродненская областная детская клиническая больница
Под агенезией (гипо-дисгенезией) понимают отсутствие мозолистых спаек, соединяющих полушария головного мозга, как частично, так и полностью.
Формирование мозолистого тела, прозрачной перегородки и свода начинается в 11 недель внутриутробной жизни и заканчивается к 16 неделе. В зависимости от этого степень дефекта мозолистого тела связана со стадией развития плода.
Первичная агенезия встречается до 12 недели жизни плода как следствие сосудистого либо воспалительного повреждения, и может быть изолированной или связанной с другими мальформациям (например: агенезия прозрачной перегородки, голопрозэнцефалия ( нарушение формирования мозга, при котором характерно полное или частичное отсутствие разделения на полушария, в сочетании с другими пороками развития ).
Вторичная дисгенезия — частичная или полная деструкция мозолистого тела —развивается уже после формирования мозолистого тела (травматические, токсические повреждения, аноксия в системе передней мозговой артерии).
Агенезия мозолистого тела чаще обнаруживается у детей с гидроцефалией, также в сочетании с другими мальформациями мозга.
Нарушение развития мозолистого тела не имеет характерной клинической картины, а неврологическая симптоматика чаще всего представлена неспецифическими интеллектуальными и неврологическими нарушениями, вплоть до полного асимптоматического течения. При сочетании агенезии мозолистого тела с другими пороками развития мозга и черепа на первый план могут выступать именно проявления последних.
КТ-признаки агенезии мозолистого тела: широко расставленные передние рога и тела боковых желудочков, высокое стояние III желудочка между ними; медиальные стенки боковых желудочков приобретают параллельный ход, а задние рога их обычно расширены (кольпоцефалия). МРТ, выполненная в 3-х плоскостях, дает полное представление о степени недоразвития мозолистого тела: частичной — гипогенезии, полной — агенезии или частичном дефекте формирования его — дисгенезии. Наиболее показательны сагиттальные срезы, позволяющие дифференцировать формирование всех его отделов. Агенезия (гипогенезия) мозолистого тела часто сочетается с отсутствием или недоразвитием гиппокампа, миндалевидного ядра, гетеротопией серого вещества или с другими мальформациями мозга.
В качестве первичного метода диагностики используют УЗИ. К сожалению, проведение процедуры эхографии в пренатальный период имеет много трудностей, в том числе из-за особенностей предлежания плода, что приводит к учащению случаев пропуска данной патологии. А при дисгенезии, гипогенезии, обнаружить дефект еще более c ложно. Затрудняет интерпретацию заболевания то, что агенезия часто сочетается с множественными пороками развития, с различными генетическими симптомами.
Агенезия мозолистого тела головного мозга на узи у новорожденного
Агенезия мозолистого тела является пороком, частота встречаемости которого и клиническая значимость неизвестны. По данным различных исследований частота его выявления варьирует и зависит от особенностей исследуемой популяции и методов диагностики.
Расчетная частота для общей популяции обычно колеблется от 0,3 до 0,7% и составляет от 2 до 3% для групп с пороками развития. Этиология заболевания гетерогенна. Возможно, важную роль в его развитии играют генетические факторы. Для него были зарегистрированы аутосомно-доминантный, аутосомно-рецессивный и сцепленный с полом типы наследования.
Агенезия мозолистого тела также является частью многих менделирующих синдромов. Высокая частота выявления сочетанных пороков при этом состоянии свидетельствует о том, что агенезия мозолистого тела является частью многих широко распространенных нарушений развития.
По данным одного из антенатальных исследований сопутствующие анатомические дефекты обнаруживались в 50% случаев и были, в основном, представлены синдромом Денди-Уокера (Dendy-Walker) и пороками сердца. Аномалии кариотипа (трисомии 18 и 8) были выявлены в 20% наблюдений.
Агенезия мозолистого тела является пороком который сопровождается минимальными анатомическими изменениями, в связи с чем его диагностика особенно до 20 нед беременности, бывает трудна даже для опытных специалистов. Развитие мозолистого тела происходит на поздних этапах церебрального онтогенеза плода, а именно: между 12 и 18 нед гестации, поэтому, вероятно, до 18 нед в большинстве случаев диагноз установить бывает невозможно.
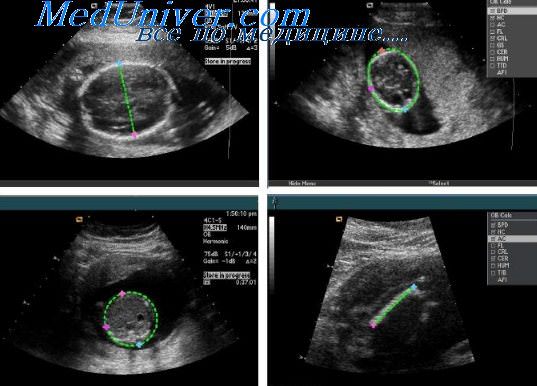
При плановых обследованиях, выполняемых позже этого срока, отсутствие возможности визуализировать полость прозрачной перегородки или расширение атриума боковых желудочков должно наводить на мысль о возможном наличии агене-зии мозолистого тела.
При подозрении на наличие этой аномалии необходимо провести поиск более специфичных признаков. Непосредственная визуализация отсутствия мозолистого тела возможна при использовании срединных фронтальной (коронарной) и продольной (сагиттальной) плоскостей сканирования. Изображения в этих плоскостях не всегда бывает легко получить, особенно при теменном предлежании плода. В таких случаях большие преимущества имеет трансвагинальная эхография.
Агенезия мозолистого тела может быть полной или частичной. В последнем случае, который называется дисгенези мозолистого тела, его каудальный отдел (комиссура и тело) отсутствует в разной степени. Полная агенезия обычно считается мальформацией, возникающей в результате нарушения эмбриогенеза, в то время как частичная может представлять собой как истинную мальформацию, так и дисрупцию, которая произошла на каком-либо сроке беременности.
Кроме того, эхографические признаки частичной агенезии еще более трудно обнаруживаются, чем при наличии полной формы. В связи с этим антенатальная диагностика такого состояния во многих случаях бывает невозможна.
Прогноз при изолированной форме агенезии мозолистого тела остается неизученным. Многие авторы полагают, что агенезия мозолистого тела не приводит к значительным последствиям для неврологического развития. Однако величина показателя специфического риска в настоящее время неизвестна.
До настоящего времении получены данные только о 30 детях с пренатально установленным диагнозом изолированной агенезии мозолистого тела (у которых отсутствовали другие аномалии и был определен нормальный кариотип), длительность постантального наблюдения за которыми варьировала от нескольких месяцев до 11 лет. Нормальное или с пограничными нарушениями неврологическое развитие было отмечено в 26 случаях (87%).